
Obsah článku
- Frynsův syndrom je vzácný genetický syndrom charakterizovaný různými vrozenými vadami. Syndrom je spojen s vývojovými abnormalitami, které mohou postihnout různé části těla, včetně obličeje, končetin, mozku, plic a srdce.
- Frynsův syndrom je dědičný a obvykle se vyskytuje sporadicky, což znamená, že není běžně zděděn od rodičů. Přesné genetické mechanismy, které vedou k tomuto syndromu, nejsou zcela známy. Diagnóza se obvykle provádí na základě klinických příznaků a vyšetření, jako je ultrazvukové vyšetření během těhotenství (prenatální diagnostika) nebo genetické testování.
- Léčba Frynsova syndromu je symptomatická a zahrnuje péči o jednotlivé vrozené vady a komplexní péči poskytovanou multidisciplinárním týmem lékařů, včetně kardiologů, genetiků, neurologů a dalších specialistů, podle potřeb jednotlivého pacienta.
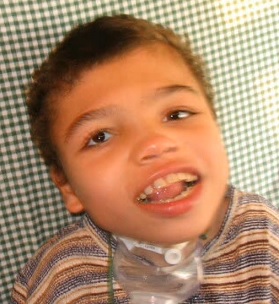 Frynsův syndrom je vzácný genetický stav, při kterém je při narození přítomno několik abnormalit. Charakteristiky syndromu jsou široce kategorizovány do bránicových vad (třeba brániční kýla) s neúplným vývojem plic, výraznými rysy obličeje, nedostatečným vývojem konců prstů na rukou a nohou (distální hypoplázie) a dalšími souvisejícími abnormalitami mozku, očí, srdce, gastrointestinální a urogenitální systém. Frynsův syndrom je považován za zděděný jako autozomálně recesivní stav, ale konkrétní kauzální gen nebo geny dosud nebyly identifikovány.
Frynsův syndrom je vzácný genetický stav, při kterém je při narození přítomno několik abnormalit. Charakteristiky syndromu jsou široce kategorizovány do bránicových vad (třeba brániční kýla) s neúplným vývojem plic, výraznými rysy obličeje, nedostatečným vývojem konců prstů na rukou a nohou (distální hypoplázie) a dalšími souvisejícími abnormalitami mozku, očí, srdce, gastrointestinální a urogenitální systém. Frynsův syndrom je považován za zděděný jako autozomálně recesivní stav, ale konkrétní kauzální gen nebo geny dosud nebyly identifikovány.
Frynsův syndrom byl poprvé popsán v roce 1979 a od té doby bylo v lékařské literatuře zaznamenáno asi 50 pacientů. I když se původně myslelo, že jde o smrtelnou poruchu, existuje i málo zdokumentovaných jedinců, kteří přežili do dětství, ačkoli přežití po novorozeneckém období je spíše výjimkou. Vzhledem k tomu, že existuje široká škála příznaků, léčba a prognóza stavu se velmi liší od člověka k člověku.
Známky a příznaky Frynsova syndromu
Frynsův syndrom je charakterizován mnoha vrozenými anomáliemi, které se u jednotlivých lidí docela liší. I když ne všichni pacienti mají následující charakteristiky, nejčastější klinické příznaky v tomto stavu jsou:
- Diafragmatické vady (týkající se bránice) a plicní hypoplázie
- Výrazný vzhled obličeje
- Distální hypoplázie
- Charakteristické anomálie: přebytečná plodová voda (polyhydramnios), zakalené rohovky a / nebo neobvykle malé oči (mikroftalmie), orofaciální štěrbina, malformace mozku, kardiovaskulární malformace, gastrointestinální malformace, renální dysplázie / renální kortikální cysty, genitální malformace
Diafragmatické vady (vady bránice)
Více než 90% jedinců s Frynsovým syndromem má vrozenou diafragmatickou kýlu (CDH): to znamená, že bránice není zcela vytvořena, což vede k tomu, že se obsah jako tenké střevo, játra a žaludek přesouvá do hrudní dutiny. Nejběžnější kýlou je jednostranná levostranná kýla. Běžně je spojen nedostatečný rozvoj plic (hypoplázie plic) a další problémy s dýcháním.
Charakteristické rysy obličeje
Jednotlivci s Frynsovým syndromem mají obvykle charakteristické rysy obličeje, jako je hrubý obličej, oči s velkým odstupem (hypertelorismus) a zakalená vnější vrstva oka (zakalená rohovka), široký a plochý nosní můstek s nosními dírkami směřujícími k horní části hlavy (anteverted nares), malformované (dysplastické) a nízko posazené uši, široká pusa (macrostomia) a malá čelist (microretrognathia). Rozštěp patra byl hlášen u 50% jedinců. Rozštěp rtu byl hlášen u 25% pacientů.
Distální hypoplázie
Jedinci s Frynsovým syndromem mohou mít nedostatečný vývoj (hypoplázii) nehtů a kostí prstů, což je hlášeno u 60% postižených. 10% pacientů má ohnuté prsty (kamptodaktylie). Bylo také popsáno zakřivení páteře ze strany na stranu (skolióza), problémy se žebry, abnormality vývoje kostí a chrupavek (osteochondrodysplazie).
Sponzorováno
Další charakteristické anomálie
U některých jedinců s Frynsovým syndromem byly hlášeny další související anomálie.
U většiny postižených dětí byly zjištěny neurologické abnormality způsobené strukturálními malformacemi mozku. Příklady abnormalit jsou ventrikulomegalie, selhání vývoje (ageneze) corpus callosum a Dandy-Walkerova malformace. Ventrikulomegalie je zvětšení postranních komor mozku, dutin, kde proudí mozkomíšní mok. Corpus callosum spojuje dvě poloviny mozku nervovými vlákny. U malformací Dandy-Walkera je zvětšena čtvrtá komora mozku a základna lebky (zadní fossa) a mozečkový vermis, oblast mozku odpovědná za koordinaci uspořádání těla, je nedostatečně rozvinutá.
Srdeční vady jsou také často u pacientů pozorovány, přičemž u 40% se jedná o defekt komorového septa, 10% o defekty síňového septa a 10% o abnormality aorty.
Vzhledem k abnormalitě bránice u pacientů někdy vyskytují další břišní defekty. Mezi ně patří omfalokéla, kde se střeva, játra a další orgány vyvíjejí mimo břišní stěnu, anální malformace a střevní malrotace, když jsou střeva zkroucená a brání průchodu potravy.
Asi 10% jedinců s Frynsovým syndromem má také abnormality genitálií a moči. Mohou být přítomny cysty v ledvinách (renální cysty). Ledviny se nemusí správně vyvíjet (renální dysplázie). Muži mohou pacienti mít jedno nebo obě varlata, které nesestoupily do šourku (kryptorchismus) nebo mají malý penis; někdy je otvor močové trubice v neobvyklé poloze (hypospadias). Ženy mohou mít abnormální dělohu ve tvaru srdce, která je rozdělena na dvě místo jedné velké dutiny nazývané bicornuate uterus.
Přežití po novorozeneckém období je vzácné a ti, kteří přežili po této fázi, mají vývojová zpoždění. Stupeň vývojového zpoždění a mentálního postižení se u jednotlivců liší. Zatímco dříve se předpokládalo, že všichni jedinci měli závažné vývojové zpoždění, bylo hlášeno několik dětí s mírnějšími poruchami učení. Růst byl hlášen jako normální u dvou dětí, ale údaje o růstu pro většinu jedinců, kteří přežili po novorozeneckém období, nejsou k dispozici.
Příčiny Frynsova syndromu
Specifické geny, které způsobují Frynsův syndrom, nejsou známy, ale kvůli vzorům detekovaným v rodinách se předpokládá, že Frynsův syndrom je autozomálně recesivní stav.
Recesivní genetické poruchy se vyskytují, když jedinec zdědí dvě kopie pozměněného genu pro stejný stav, jednu od každého rodiče. Pokud jedinec zdědí jeden normální gen a jeden gen pro tuto nemoc, bude osoba nositelem této nemoci, ale obvykle nebude vykazovat příznaky. Riziko, že dva rodiče, kteří jsou nositeli, projdou změněným genem a budou mít postižené dítě, je 25% s každým těhotenstvím..
Léčba Frynsova syndromu
V současné době neexistuje lék na Frynsův syndrom. Léčba je zaměřena na řešení potřeb jednotlivce. Často se doporučuje chirurgický zákrok na brániční kýlu a / nebo podpůrná opatření.
Vzhledem k život ohrožující povaze určitých komplikací, jako je bránicová kýla, nedostatečný vývoj plic a srdeční vady, mohou být u novorozence nutné zákroky už krátce po narození. Například děti s bránicovou kýlou jsou intubováni, aby se zabránilo nafouknutí herniovaného střeva. Může být vhodná konzultace dětských neurologů, kardiologů, gastroenterologů, nefrologů a kraniofaciálních specialistů.
Sponzorováno
Rodičům dítěte s Frynsovým syndromem se doporučuje genetické poradenství.
Zdroje článku a studie o Frynsově syndromu
- Studie o Frynsově syndromu Autor: Anne Slavotinek, MBBS, PhD
- CONGENITAL DIAPHRAGMATIC HERNIA ASSOCIATED WITH FRYNS SYNDROME Autor: Surekha Arakeri
Sponzorováno
Autor článku
Líbil se vám náš článek? Sdílejte ho, uděláte nám radost
Štítky: Genetická onemocnění, Vzácná onemocnění
Přečtěte si také naše další články