
Obsah článku
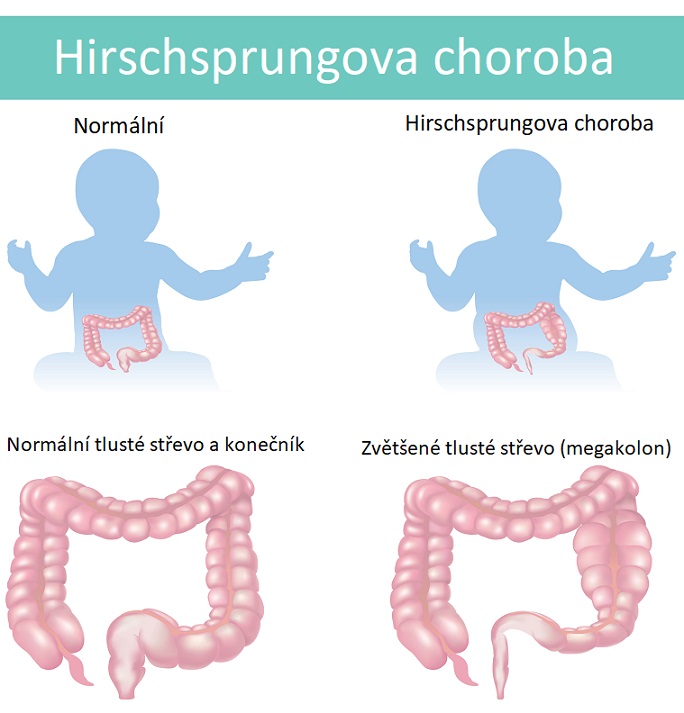
- Hirschsprungova choroba je stav, který postihuje tlusté střevo a způsobuje problémy s průchodem stolice. Hirschsprungova choroba je vzácné onemocnění tlustého střeva, které postihuje trávicí systém novorozenců, kojenců a malých dětí. Tato porucha vzniká v důsledku nepřítomnosti nervových buněk (gangliových buněk) v určité části střeva, což má za následek neschopnost normálního stahování a přenosu střevního obsahu (stolice) směrem k řitnímu otvoru.
- Tento stav je přítomen při narození (je tedy vrozený) v důsledku chybějících nervových buněk ve svalech tlustého střeva dítěte.
- Novorozenec, který má Hirschsprungovu chorobu, obvykle nemůže vyloučit stolici už v prvních dnech po narození.
- V mírných případech nemoci může být stav zjištěn až později v dětství. Méně často je Hirschsprungova choroba poprvé diagnostikována u dospělých.
- Léčba je často provedena chirurgickým zákrokem k obejití nebo odstranění nemocné části tlustého střeva. Záleží ale případ od případu. Léčba zahrnuje chirurgický zákrok, během kterého je postižená část tlustého střeva odstraněna. Po chirurgickém zákroku může být nutná následná péče a sledování. Časná diagnóza a léčba jsou klíčové pro minimalizaci komplikací a zlepšení dlouhodobé prognózy.
Hirschsprungova choroba (HSCR) je vrozená vada. Tato porucha je charakterizována absencí určitých nervových buněk (ganglií) v segmentu střeva u kojence. Absence gangliových buněk způsobuje, že svaly ve střevech ztrácejí schopnost pohybovat stolicí střevem (je tedy výrazně narušena peristaltika).
Peristaltika je normální proces těla. Peristaltika vytváří vlnové kontrakce ve svalech lemujících střeva. Tyto kontrakce pohánějí stolici a další odpadní materiál trávicí soustavou. Neúčinná peristaltika vede k podpoře stolice ve střevech. U postižených jedinců se může objevit zácpa a částečná nebo úplná obstrukce střev. Může dojít k velké bolesti a pocitu nepohodlí.
Pokud tento problém nebude léčen, může se vyvinout potenciálně závažná bakteriální infekce. Specifické příznaky se mohou u jednotlivých osob hodně lišit. Hirschsprungova choroba (HSCR) se může objevit jako izolovaný problém nebo jako součást nějaké širší poruchy, která postihuje více orgánových systémů.
Hirschsprungova choroba – ilustrace
Příznaky Hirschsprungovy choroby
Mezi příznaky v novorozeneckém období patří neschopnost mekonia (první stolice novorozence) projít střevem v krátké době po narození. Mekonium je tmavá lepkavá látka, která je obvykle přítomna ve střevě při narození a je vylučována při prvním pohybu střev po narození dítěte. Pokud neprojde první stolice po dobu 24-48 hodin, může to naznačovat Hirschsprungovu chorobu.
Kojenci s Hirschsprungovou chorobou budou mít velmi často otoky, trpí bolestí břicha a zvracením. Postižení kojenci mají zácpu a často vykazují špatný přírůstek hmotnosti a pomalý růst.
Hirschsprungova choroba může někdy vést k onemocnění nazývanému enterokolitida, což je zánět tenkého střeva a tlustého střeva. Toto se často označuje jako enterokolitida spojená s Hirschsprungem. Enterokolitida spojená s Hirschsprungem je nejčastější komplikací této choroby vyskytující se u 30-40% jedinců s touto nemocí a může mít mírnou až těžkou povahu. Enterokolitida spojená s Hirschsprungem se často projevuje horečkou, výbušným průjmem, otokem břicha, letargií a zvracením. U některých jedinců s těžkou nebo neléčenou enterokolitidou spojenou s Hirschsprungem se může vyvinout sepse, což je rozšířená bakteriální infekce krevního řečiště a je potenciálně život ohrožující. Těžká nebo neléčená enterokolitida může také vést k toxickému megakolonu, další život ohrožující komplikaci. Toxické megakolon je obávanou komplikací chorob střev a ohrožuje pacienta akutně na životě svými komplikacemi. Jedinec s Hirschsprungovou chorobou, u kterého se tyto příznaky objeví, by měl samozřejmě naléhavě vyhledat lékařskou pomoc.
Přibližně 90% počátečních diagnóz Hirschsprungovy choroby je stanoveno během prvního roku života. Většina ze zbývajících 10% se objeví v raném dětství, méně než 1% se tvoří v dospívání nebo dospělosti. Není divu, že tito jedinci často uvádějí celoživotní problém se zácpou.
Příčiny Hirschsprungovy choroby
Hirschsprungova choroba, která se vyskytuje jako izolovaný problém, byla spojena s mutacemi v několika různých genech. Přibližně 50% postižených jedinců má jednu z těchto genových abnormalit. Tyto genové změny způsobují, že lidé jsou náchylní k rozvoji poruchy. Osoba, která má genetickou predispozici k poruše, nese gen (nebo geny) pro toto onemocnění, ale nemusí být projevena, pokud není spuštěna nebo „aktivována“ za určitých okolností, například v důsledku zvláštních faktorů prostředí (multifaktoriální dědičnost).
Dědičnost těchto genových změn může být dominantní nebo recesivní v závislosti na příslušném genu, ale pro vznik poruchy je pravděpodobně nutná přítomnost více abnormálních genů. Abnormální geny zapojené do Hirschsprungovy choroby mohou mít různé účinky na členy stejné rodiny. Pokud mají rodiče postižené dítě, zvyšuje se jejich šance na další dítě s touto poruchou. Rodič, který má Hirschsprungovu chorobu, má také větší šanci mít dítě s touto poruchou.
Geny spojené s touto nemocí jsou ve dvou hlavních skupinách nazývaných RET geny a EDNRB geny. Když porucha ovlivňuje krátký segment tlustého střeva, hlavním zapojeným genem je gen RET umístěný na chromozomu 10q11.2.
Sponzorováno
Když dojde k Hirschsprungově chorobě spolu s dalšími abnormalitami, příčinou je často abnormalita chromozomu nebo genetický syndrom. Třeba lidé s Downovým syndromem mají větší riziko vzniku Hirschsprungovy choroby než lidé v běžné populaci. Mezi genetické syndromy, které mohou být spojeny s Hirschsprungovou chorobou, patří Mowat-Wilsonův syndrom, Waardenburgův syndrom, Bardet-Biedelův syndrom, syndrom hypoplastických chrupavek a vlasů a vlasů, syndrom vrozené centrální hypoventilace, Frynsův syndrom, mnohočetná endokrinní neoplazie typu 2, Smithův-Lemliův-Opitzův syndrom, syndrom L1 a Pitt-Hopkinsův syndrom.
Známky a příznaky Hirschsprungovy choroby (HSCR) se objevují v důsledku selhání vývoje specifických nervových buněk zvaných gangliony v části tlustého střeva kojence. Vzhledem k tomu, že ve střevě chybí gangliony, nelze stolici tlačit střevem ven z těla pomocí peristaltiky.
Délka střev, která je ovlivněna touto nemocí, se může lišit. U přibližně 80% postižených kojenců je postiženo tlusté střevo. Konečník je poslední část tlustého střeva a spojuje konečník s sigmoidním tlustým střevem. U kojenců s absencí gangliových buněk v konečníku a sigmoidním tlustém střevě se říká, že mají „krátký segment“ Hirschsprungovy choroby. Zatímco přibližně 12% kojenců bude mít gangliové buňky chybějící ve většině tlustého střeva a bude označováno jako „Hirschsprungova choroba s dlouhým segmentem“, a přibližně 7% bude mít gangliové buňky chybějící v celém tlustém střevě.
Diagnostika
Diagnóza Hirschsprungovy choroby může být provedena na základě fyzického vyšetření, kompletní pacientovi a rodinné anamnézy, identifikace charakteristických příznaků a řady specializovaných testů. Většina lidí (85-90%) je diagnostikována hned v raném dětství.
Prvním příznakem je obvykle neschopnost mekonia projít střevy. Preferovaným diagnostickým testem je sací biopsie konečníku. Biopsie zahrnuje chirurgické vyříznutí malého vzorku postižené tkáně a její studium pod mikroskopem. Absence gangliových buněk potvrzuje diagnózu.
Mezi další testy, které lze použít, patří rentgen břicha, který odhalí přítomnost střevní blokády, anorektální manometrie, která zahrnuje použití balónků a tlakových senzorů k posouzení zdraví a funkce konečníku a kontrastní nebo bariový klystýr, který zahrnuje použití kontrastní látky v konečníku. Kontrastní látka je látka, která se používá ke zlepšení vzhledu struktury nebo části těla na rentgenovém snímku. Po použití kontrastní klystýru v konečníku se provádí rentgenové záření k posouzení zdraví a funkce tlustého střeva.
Anorektální manometrie je specializovaná metoda, která umožňuje vyšetřit činnost análních svěračů a určit tlak ve vnitřním i zevním svěrači konečníku.
Pokud jsou kromě Hirschsprungovy choroby přítomny i jiné abnormality, je možné, že je nemoc způsobena chromozomální abnormalitou nebo genetickým syndromem. Jedinci s vícečetnými anomáliemi by měli být vyšetřeni genetikem, aby se pokusil stanovit základní diagnózu.
Léčba Hirschsprungovy choroby
Téměř ve všech případech vyžaduje léčba nějaký chirurgický zákrok k odstranění části tlustého střeva a / nebo konečníku, která postrádá normální vývoj nervů – pak může dojít spojení obou zdravých konců. Existují tři standardní chirurgické postupy určené k nápravě této poruchy. Volba postupu závisí na názoru a zkušenostech chirurga. Každý postup odstraní postiženou část a připojí zdravou část střeva ke konečníku. V současné době se většina postupů provádí v jedné fázi.
Pokud se dítě narodí předčasně, má nízkou porodní váhu nebo pokud je kriticky nemocné, může chirurg rodičům poradit, že bezpečnější přístup je vícestupňová strategie. První fází je vytvoření dočasné kolostomie, při které se zdravé koncové střevo před postiženým střevem dostane na povrch břicha a vytvoří stomii. Tímto otvorem neboli „stomií“ se obsah střev vyloučí do speciálního vaku a odstraní se. Po určité době se provede druhá fáze operace, kdy může být stomie uzavřena. Většina dětí s Hirschsprungovou chorobou (HSCR) nepotřebuje kolostomii ani ileostomii.
Podle lékařské literatury si většina dětí po úspěšné operaci užívá dobré až vynikající kvality života. Ve vzácných případech mohou některé děti vyžadovat revizní nebo opakovanou operaci.
Genetické poradenství může být přínosem pro postižené jedince a jejich rodiny.
VIDEO: Neočekávaná komplikace po operaci Hirschprungovy choroby a její řešení
Sponzorováno
Zdroje článku a studie
- Hirschsprung's Disease: a Clinical and Pathologic Study in Iranian Constipated Children Autor: Maryam Monajemzadeh, MD
- A genetic study of Hirschsprung disease Autor: J A Badner
- Zdroj obrázku ve článku: pattarawit / depositphotos.com
Sponzorováno
Autor článku
Líbil se vám náš článek? Sdílejte ho, uděláte nám radost
Štítky: Dětské nemoci, Genetická onemocnění, Střeva
Přečtěte si také naše další články